Lungenhochdruck (PAH)
Was ist das?
Die pulmonale Hypertonie (PH) ist häufig Ausdruck einer schweren linkskardialen oder pulmonalen Grunderkrankung. Nur selten kommt es zu dem Nachweis eines primären vaskulären Umbaus der Lungenarterien – der pulmonal-arteriellen Hypertonie (PAH). Obschon wir in den letzten Jahren pathophysiologische Mechanismen der PAH zunehmend besser verstehen, sind Auslöser der Erkrankung unverändert nicht endgültig definiert. Auch therapeutische Interventionen beschränkten sich abseits von präklinischen und einigen klinischen Studien vorwiegend auf eine Vasodilatation im pulmonalen Gefäßstrombett. Erfreulicherweise können wir hier jedoch ein immer größer werdendes Portfolio aus Medikamenten einsetzen, welches individuell auf den Patienten abgestimmt eine relevante Verbesserung der Morbidität und Mortalität bedeutet. Diagnostik und Therapie der PH sollten weiterhin an Zentren konzentriert werden, um eine bestmögliche Versorgung Betroffener gewährleisten zu können. Die PH-Zentren in der Region sind eng im PH-Netzwerk Nordrhein miteinander verknüpft und treffen sich mehrmals jährlich zur kritischen Diskussion aktueller Entwicklungen.
2018 hat die hämodynamische Definition der PH aufgrund neuerer Erkenntnisse erhebliche Veränderungen erfahren. Diese wurden und werden nicht nur in der PH-Community kritisch diskutiert. Der vorliegende Beitrag soll Vor- und Nachteile der neuen PH-Definition beleuchten und aktuelle Therapieoptionen abhängig von der Risikoklasse darstellen.

Diagnostik
Klinische Klassifikation der PH
Die PH ist eine Erkrankung, deren Diagnose erst auf Basis einer hämodynamischen Diagnostik im Rahmen einer Rechtsherzkatheteruntersuchung definitiv gestellt werden kann. Der Verdacht einer PH erhärtet sich im Alltag jedoch oft bereits vor Durchführung einer invasiven hämodynamischen Diagnostik. Hierbei können die Rechtsachsenabweichung im Elektrokardiogramm, eine belastungsinduzierte Dyspnoe, Unterschenkelödeme oder die Halsvenenstauung genauso wie eine Erhöhung des N-terminalen pro brain natriuretic peptid (NT-proBNP) wegweisend sein.
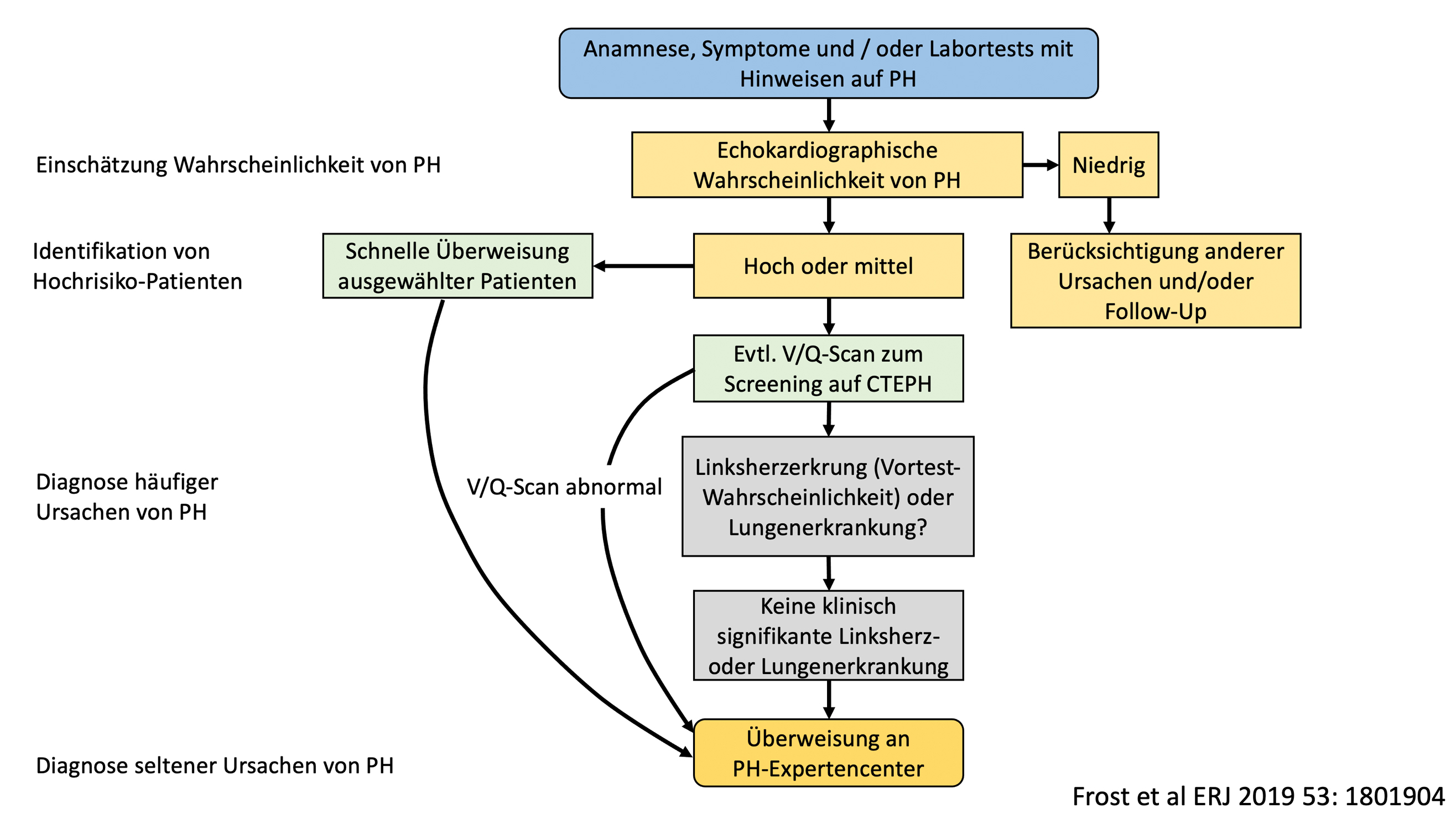
Zentrales Diagnostikum zur Evaluation der Wahrscheinlichkeit einer vorliegenden PH und Differentialdiagnostik ist unverändert die transthorakale Echokardiographie. Sollte es hierbei zu direkten oder indirekten Hinweisen auf das Vorliegen einer PH kommen, empfiehlt sich die Einleitung weiterer diagnostischer Schritte oder im Falle eindeutiger Hinweise auf das Vorliegen einer PH die unmittelbare Zuweisung in ein PH-Expertenzentrum. Abbildung 1 gibt einen diagnostischen Algorithmus angelehnt an den Vorschlag der internationalen Arbeitsgruppe „Diagnosis of pulmonary hypertension“ des 6th World Symposium on Pulmonary Hypertension vor. Neben einer Triskupidalklappenregurgitationsgeschwindigkeit >2,8 m/s sind während einer Echokardiographie auch indirekte Zeichen für das Vorliegen einer PH von großer Bedeutung. Hierzu zählen die Abflachung des Septums, die Dilatation des rechten Ventrikels und Atriums, der vergrößerte Durchmesser der Pulmonalarterie und der Vena cava inferior sowie die Myokardhypertrophie des rechten Ventrikels mit eingeschränkter rechtsventrikulärer Pumpfunktion.
Im Falle des Verdachts einer PH auf Basis der echokardiographischen Diagnostik sollte anschließend das Vorliegen einer relevanten Linksherzerkrankung, Lungenerkrankung oder thromboembolischen Lungenerkrankung untersucht werden. Unverändert sind hier die Computertomographie des Thorax, die Ventilations-Perfusionsszintigraphie und eine lungenfunktionelle Diagnostik inklusive Bodyplethysmographie und Messung der Diffusionskapazität wegweisend. Auch die Durchführung einer Spiroergometrie hat sich in den letzten Jahren als hilfreiches Diagnostikum erwiesen. Hierbei kann nicht nur die globale kardiopulmonale Leistungsfähigkeit beurteilt, sondern auch auf Basis des Atemäquivalents für Kohlendioxid unmittelbar die Wahrscheinlichkeit einer pulmonalen Gefäßerkrankung abgeschätzt werden. Das Atemäquivalent für Kohlendioxid beschreibt wieviel Liter Luft ein Mensch atmen muss, um einen Liter Kohlendioxid abzuatmen und ist im Falle erhöhter Werte ein sensitiver Marker für das Vorliegen einer PH.
Ergibt sich die Indikation zur Durchführung eines Rechtsherzkatheters, sollte dieser möglichst in einem PH-Zentrum durchgeführt werden, um eine vollständige Diagnostik mit präziser Messung aller wichtigen hämodynamischen Parameter und etwaiger ergänzender Durchführung einer Vasoreagibilitätstestung und ggf. eines sogenannten „Volume challenge“ sicherzustellen. Zu den obligat zu erhebenden Parametern gehört neben der Druckmessung eine Reihe weiter Meßwerte (s.u.). Nur eine vollständige hämodynamische Diagnostik erlaubt am Ende eine sichere Diagnose und die bestmögliche Therapie für jeden einzelnen Patienten.
Die klinische Klassifikation der PH beruht auf der Einteilung nach Nizza (Tabelle 1). Grob unterscheidet man 5 Gruppen, die entsprechend der Ätiologie der PH gruppiert sind. Gruppe 1 umfasst alle Entitäten, die wir unter dem Oberbegriff PAH zusammenfassen. Hierunter fallen die idiopathische sowie hereditäre PAH, genauso wie PAH im Rahmen einer Sklerodermie, HIV-Infektion oder Schistosomiasis (assoziierte Formen). Seit 2018 ist in Gruppe 1 auch die PAH, die langfristig auf eine hochdosierte Kalziumantagonistentherapie anspricht, als eigene Entität aufgeführt. Gruppe 2 umfasst all jene PH-Formen, die auf eine Linksherzerkrankung zurückzuführen sind. Neben der Linksherzinsuffizienz mit erhaltener und reduzierter linksventrikulärer Pumpfunktion sind auch valvuläre Ursachen oder Kombinationen aus z.B. Herzinsuffizienz und funktioneller Mitralinsuffizienz mit zu berücksichtigen. In Gruppe 3 sind Krankheiten der Lunge aufgeführt, die zu einer PH führen können. Lange wurde darüber diskutiert, ob auch die Sarkoidose neben den obstruktiven und restriktiven Lungenerkrankungen der Klasse 3 zuzuordnen ist, am Ende entschied man sich jedoch dagegen und ordnete die Sarkoidose weiterhin der Klasse 5 zu. Klasse 4 besteht im Wesentlichen aus der chronisch thromboembolischen PH. Diese bezeichnet einen Residualzustand, den manche Patienten nach einer Lungenembolie erleiden und der unbehandelt eine schlechte Prognose aufweist. Klasse 5 umfasst schließlich alle Erkrankungen mit unklarem oder multifaktoriellem Mechanismus.
Eine sichere PH-Diagnose erfordert immer einen Rechtsherzkatheter und eine breite Differentialdiagnostik der Genese.
Definition
Hämodynamische Definition der PH
Bis zum Dezember des Jahres 2018 war die Definition der pulmonalen Hypertonie maßgeblich an einen pulmonal arteriellen Mitteldruck (PAPm) 25 mmHg gebunden. Auf dem 6. Weltsymposium zur PH in Nizza, deren Manuskripte kürzlich publiziert wurden, wurden nun andere Grenzwerte vorgeschlagen. Die präkapilläre PH ist demnach definiert als PAPm > 20 mmHg mit einem pulmonalkapillären Verschlussdruck (PAWP) ≤ 15 mmHg und einem pulmonalvaskulären Widerstand (PVR) von 3 WU (Tabelle 2). Letzterer ergibt sich aus dem Quotienten von PAPm-PAWP-Differenz und dem Herzzeitvolumen (HZV). Eine präkapilläre PH finden wir bei Patienten mit Lungenerkrankungen, der chronisch-thromboembolischen PH, aber auch der PAH. Erkrankungen des linken Herzens sind typischerweise durch eine Erhöhung des PAWP > 15 mmHg charakterisiert, es liegt also primär eine passive Druckerhöhung durch Rückstau erhöhter Füllungsdrucke aus dem linken Herzen eine postkapilläre PH mit sekundärer Hochregulation des Druckniveaus im präkapillären Gefäßstrombett vor. Sollte der PVR in dieser Konstellation <3WU liegen, spricht man von einer isolierten postkapillären PH, bei einem PVR 3WU hingegen von einer kombiniert prä- und postkapillären PH (CpcPH).
Maßgeblich beruht die Erneuerung der hämodynamischen Definition auf zwei wichtigen Erkenntnissen. Man weiß inzwischen sehr genau, dass der obere Grenznormbereich für den PAPm bei 20 mmHg liegt. Kovacs und Kollegen sichteten im Jahre 2009 47 Studien, um im kleinen Kreislauf hämodynamische Normalwerte zu definieren. Unabhängig der Ethnie und des Geschlechts lag der PAPm bei 13,8 ± 3,1 mmHg. Bei der Annahme dieses Mittelwertes errechnet sich daher ein oberer Grenzwert für Normalität von 20 mmHg (Mittelwert + 2 Standardabweichungen). In mehreren Studien konnte zudem jüngst eine signifikante Erhöhung der Mortalität bei Patienten mit einer moderaten pulmonalen Druckerhöhung (PAPm zwischen 21 und 25 mmHg) nachgewiesen werden.
Bedauerlicherweise bringen diese Erkenntnisse ein gewisses Dilemma mit sich, da wir nun auf der einen Seite um die Bedeutung der leichten pulmonalen Druckerhöhung wissen, sämtliche zulassungsrelevanten PAH-Therapiestudien jedoch einen PAPm 25 mmHg in den Einschlusskriterien verankert hatten. Bis zu dem Zeitpunkt, an dem ein additiver Benefit durch eine gezielte Gefäßtherapie in klinischen Studien auch bei einer leichten pulmonalen Druckerhöhung gezeigt wurde, muss daher für die Indikation zur gezielten PAH-Therapie weiterhin ein Grenzwert von 25mmHg gelten. Eine Ausnahme stellen möglicherweise Patienten mit Sklerodermie dar, bei denen in präliminären Daten ein Benefit von einer gezielten Gefäßtherapie gezeigt werden konnte. Fest steht, Patienten mit einem PAPm zwischen 21 und 24 mmHg sollten engmaschig kontrolliert werden, insbesondere im Falle systemischer Erkrankungen, die mit einer PH assoziiert sind.
Ein PVR ≥3WU kann bereits ab einem PAPm > 20 mmHg Hinweis auf eine pulmonal-vaskuläre Erkrankung sein.
Risikostratifizierung
Und medikamentöse Therapieoptionen bei PAH
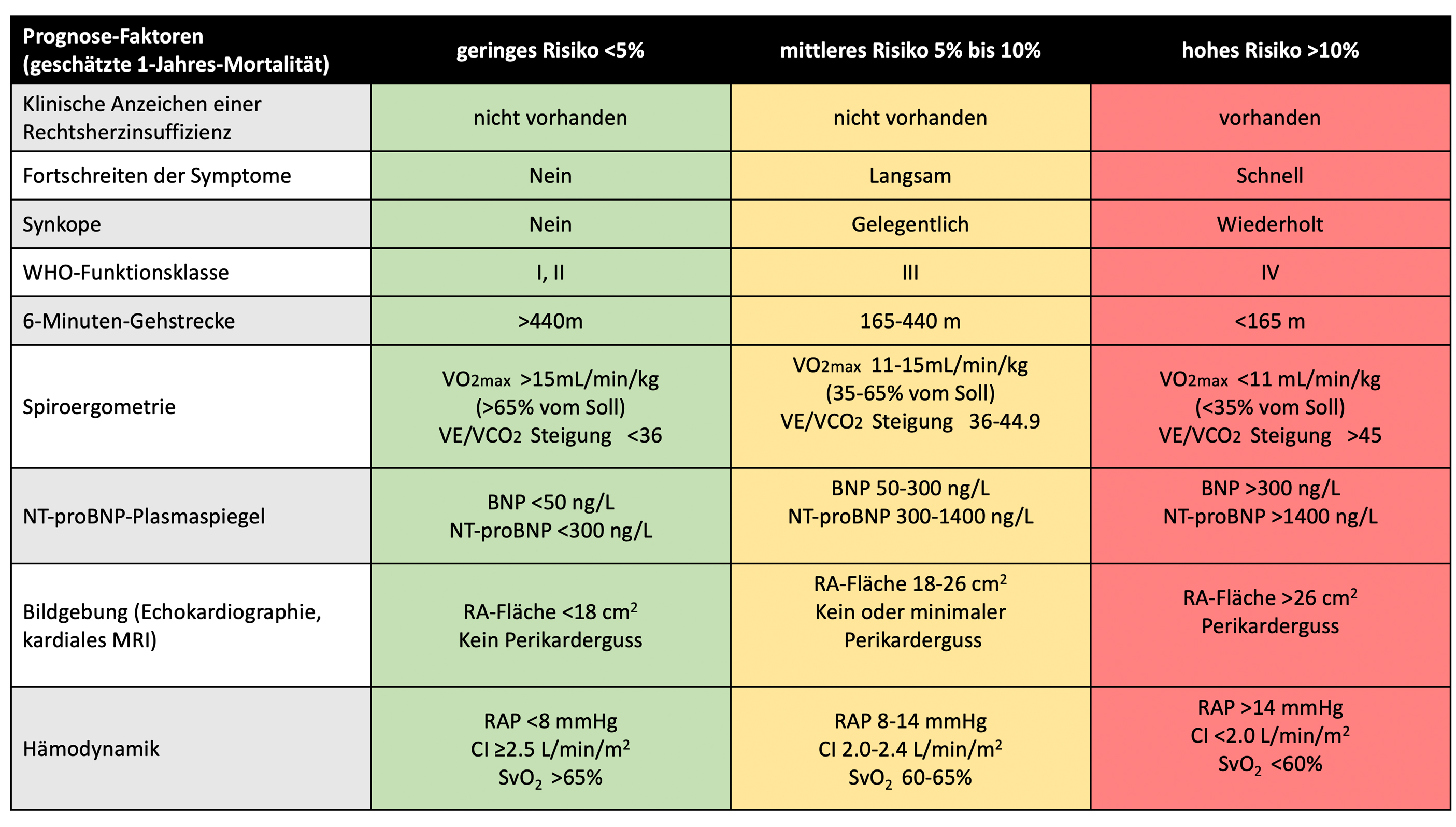
Nachdem die ESC/ERS-PH-Leitlinien im Jahre 2015 ein damals noch nicht validiertes Schema zur Risikostratifizierung von Patienten mit PAH vorgeschlagen hatten, erfuhr dieses Konzept in den letzten Jahren eine intensive wissenschaftliche Auseinandersetzung (Tabelle 3). Mehrere PH-Register (SPAHR, FPHN, COMPERA) in Europa bestätigten, dass die vorgeschlagene Risikostratifizierung sehr gut das individuelle Mortalitätsrisiko eines Patienten widerspiegelt. Daher ist dieses Schema heute Basis, um über das strategisch-therapeutische Vorgehen zu entscheiden. Man unterscheidet 3 Risikoklassen, die das 1-Jahres-Mortalitätsrisiko abschätzen. Gruppe 1 hat ein Mortalitätrisiko < 5%, Gruppe 2 hat ein intermediäres Risiko mit 5-10% und Gruppe 3 ein hohes Risiko mit einer 1-Jahresmortaltität >10%. Prognostisch bedeutsame Parameter umfassen klinische Zeichen einer Rechtsherzinsuffizienz, eine Symptomprogression, die 6-Minuten-Gehstrecke sowie die globale kardiopulmonale Leistungsfähigkeit auf Basis der Spiroergometrie, des NT-proBNP Level sowie echokardiographische und hämodynamische Parameter. In Abhängigkeit der Risikoklasse wird über das therapeutische Vorgehen entschieden. Während man bei einem niedrigen oder intermediärem Risiko heute in der Regel mit einer oralen Kombinationstherapie beginnt, erfordert das hohe Risiko eine unmittelbare 3-fach-Kombinationstherapie inklusive eines intravenösen Prostanoids.
Grundsätzlich stehen therapeutisch heute verschiedene orale, inhalative, subkutane sowie intravenöse Substanzen zur Senkung des pulmonal arteriellen Drucks zur Verfügung. Diese adressieren verschiedene biochemische Signalwege und führen zu einer Vasodilatation und Hemmung der Zellproliferation in der Gefäßwand. 3 Signalwege können wir heutzutage therapeutisch adressieren:
- Phosphodiesterase-5-(PDE5)-Inhibitoren (Sildenafil, Tadalafil) induzieren durch die Hemmung des Abbaus von zyklischem Guanosinmonophosphat (cGMP) eine Vasodilatation im pulmonalen Gefäßbett. Einen ähnlichen Effekt bewirkt auch der sGC-Stimulator Riociguat, nicht jedoch über die Hemmung des Abbaus von cGMP, sondern durch eine Stimulation der cGMP-Produktion. Phosphodiesterase-5-Inhibitoren und Riociguat dürfen aufgrund der ähnlichen Wirkweise nicht kombiniert werden.
- Endothelin-Rezeptor-Antagonisten (ERA) (Ambrisentan, Bosentan, Macitentan) wirken über eine Blockade des Endothelinrezeptors einer Vasokonstriktion und Proliferation entgegen.
- Prostazyklin-Analoga wie Epoprostenol, Iloprost und Treprostinil induzieren über ihre Wirkung am Prostazyklinrezeptor eine Erhöhung von zyklischem Adenosinmonophosphat und darüber eine konsekutive Vasodilatation. Mit dem Prostazyklin-Rezeptor-Agonisten Selexipag ist seit kurzem auch eine orale Substanz in Deutschland zugelassen, die durch eine agonistische Wirkung am Prostazyklinrezeptor ähnliche Effekte aufweist.
Zu den großen klinischen, randomisierten, kontrollierten Studien der letzten Jahre, die 2- und 3-fach-Kombinationstherapien mit einer Monotherapie beziehungsweise 2-fach Kombinationstherapie verglichen, gehören SERAPHIN, AMBITION und GRIPHON. Diese Studien hatten im Gegensatz zu den bis dato existierenden Studien, die vor allem eine Verbesserung der 6-Minuten-Gehstrecke untersuchten, allesamt kombinierte Mortalitäts- und Morbiditäts-Endpunkte. Alle 3 Studien wiesen signifikante Vorteile einer Kombinationstherapie gegenüber der Gabe von Einzelsubstanzen auf.
Typischerweise würde man daher heutzutage bei einem Patienten mit einer neu diagnostizierten PAH initial mit einer Kombinationstherapie aus einem ERA und einem PDE5-Inhibitor starten. Alternativ kann man den ERA auch mit Riociguat kombinieren. Zudem gibt es erste Hinweise darauf, dass auch Patienten mit einer bereits bestehenden Kombinationstherapie aus einem ERA und einem PDE-5-Hemmer von einem Austausch des PDE-5-Hemmers gegen Riociguat profitieren können (RESPITE-Studie). Eine aktuell durchgeführte randomisierte, kontrollierte Multicenterstudie (REPLACE) soll diesbezüglich weitere Klarheit bringen.
Sollten Patienten durch diese Kombinationstherapie nicht in die niedrige Risikoklasse wechseln, so kann eine weitere Eskalation der Therapiemaßnahmen indiziert sein. Im Falle einer WHO-Funktionsklasse II oder III besteht die Option, den oral verfügbaren Prostazyklinrezeptoragonisten Selexipag einzusetzen. Sollte trotz der Kombinationstherapie gar ein hohes 1-Jahresmortalitätsrisiko oder die WHO-Funktionsklasse IV vorliegen, besteht die Indikation zur parenteralen Prostanoidtherapie. Diese Therapie kann subcutan oder intravenös über dauerhaft liegende Katheter (z.B. Hickman Katheter) oder subcutan implantierte Pumpen (z.B. LenusPro-Pumpe) verabreicht werden. Iloprost liegt auch in der inhalativen Form vor und kann bei Patienten eingesetzt werden, die sich für eine dauerhafte intravenöse Therapie nicht eignen.
Nicht vergessen sollte man im Rahmen einer intensiveren vasospezifischen Therapie auch die Zunahme potenzieller Nebenwirkungen. Die Kölner Konsensus Konferenz empfiehlt daher bei Patienten mit relevanten Komorbiditäten wie arterieller Hypertonie, koronarer Herzkrankheit, Diabetes mellitus, Adipositas u.a. durchaus zunächst die Verträglichkeit der Therapie in Form einer gezielten Monotherapie zu prüfen, um der Gefahr von Nebenwirkungen entgegenzuwirken. Keinesfalls sollte dies jedoch dazu führen, dass potenziell lebensverlängernde Kombinationstherapien im Verlauf nicht erwogen werden.
Bei hohem Mortalitätsrisiko ist eine frühe Kombinationstherapie inklusive intravenöser Prostanoide entscheidend.
Therapie
Supportive Therapiemaßnahmen
Neben der gezielten medikamentösen Therapie haben auch adjuvante Therapiemaßnahmen einen hohen Stellenwert und sollten im Gesamtkonzept der Behandlung der PAH nicht vergessen werden. Allen voran sind diesbezüglich supervidierte Rehabilitationsmaßnahmen zu nennen, die idealerweise in einer Einrichtung angegliedert an ein PH-Expertenzentrum durchgeführt werden. Gezieltes Training ist in der Lage, die globale kardiopulmonale Leistungsfähigkeit substanziell zu steigern sowie die 6-Minuten-Gehstrecke und die Lebensqualität zu verbessern.
Diuretika und hier insbesondere die Gabe des Aldosteronantagonisten Spironolacton sind essentiell in der Senkung der Vorlast und sollten bei allen Patienten mit PAH erwogen werden. Auch die intravenöse Gabe von Eisen hat sich im Falle eines Eisenmangels genau wie bei der Linksherzinuffizienz als vorteilhaft erwiesen. Die Schwangerschaft sowie die Vollnarkose weisen unverändert ein nicht unerhebliches Mortalitäts-Risiko auf. Beides sollte bei Patienten mit PAH daher möglichst vermieden werden. Ob PAH-Patienten tatsächlich rigoros vom Fliegen abgeraten werden muss, wie aktuell empfohlen, untersucht die sogenannte Pegasus-Studie. Hier werden seit 2017 aktiv Patienten rekrutiert und auch weiterhin gesucht.
Ähnlich wie bei anderen Risikokollektiven ist auch bei PAH-Patienten eine Influenza- und Pneumokokkenimmunisierung empfohlen. Außerdem sollte im Falle einer chronischen Hypoxämie mit einem arteriellen Sauerstoffpartialdruck < 60mmHg eine Langzeitsauerstofftherapie eingeleitet werden.
Überwachtes körperliches Training verbessert die globale kardiopulmonale Leistungsfähigkeit und Lebensqualität.
PH-Netzwerk Nordrhein
Pulmonale Hypertonie in der Region
Ursachen einer PH sind mannigfaltig und erfordern daher ein hohes Maß an Interdisziplinarität als auch die Kooperation von PH-Zentren untereinander. In der Region besitzt das „PH-Netzwerk Nordrhein“, in dem unter Führung des überregionalen PH-Zentrums Köln zahlreiche Institutionen angeschlossen sind, Modell-Charakter. In Köln und Solingen kooperieren Kardiologen, Pneumologen, Rheumatologen, Gastroenterologen Radiologen, Infektiologen und Humangenetiker, um den Anforderungen dieses interdisziplinären Krankheitsbildes gerecht zu werden. Während in der Uniklinik Köln die entsprechenden Fakultäten einen engen inhaltlichen Austausch gewährleisten, existiert in Solingen seit 2008 das „Forum Lungenhochdruck Solingen“. Hier treffen sich niedergelassene sowie Krankenhausärzte aus der Region quartalsweise zu einem interkollegialen Austausch und der Diskussion spezieller Krankheitsverläufe. Dieser regelmäßige Austausch ermöglicht auch im klinischen Alltag eine enge Zusammenarbeit mit dem Anspruch einer bestmöglichen Patientenversorgung.
Fazit
Das 6. Weltsymposium PH in Nizza brachte im Jahre 2018 einige Neuerungen im Hinblick auf hämodynamische Definitionen und Therapie-Empfehlungen der PAH mit sich. Was die neuen Grenzwerte der PH-Definition im klinischen Alltag am Ende tatsächlich bedeuten, bleibt jedoch abzuwarten. Der medikamentösen Kombinationstherapie kommt bei PAH eine immer bedeutendere Rolle zu. Hierzu zählt insbesondere auch die parenterale Prostanoidtherapie, die häufig noch zu spät erwogen wird. Auch supportive Therapiemaßnahmen verbessern die Prognose, Lebensqualität und globale kardiopulmonale Leistungsfähigkeit und stellen daher eine bedeutsame Säule im therapeutischen Gesamtkonzept dar. Wichtig ist weiterhin die kritische Auseinandersetzung mit den individuellen Anforderungen eines jeden Patienten. Dabei spielen Komorbiditäten genauso wie persönliche Wünsche des Patienten eine gewichtige Rolle.
Die Behandlung der PAH bleibt auch zukünftig eine Herausforderung. Dank medikamentöser Innovationen haben wir jedoch die Möglichkeit aus einem immer größer werdenden therapeutischen Portfolio für jeden Patienten die bestmögliche Therapie individuell zusammenzustellen, um Mortalität und Mortalität zu senken und die Lebensqualität zu verbessern.
Diagnostik und Therapie der PAH sollten auch zukünftig an Zentren mit interdisziplinärer Expertise gebündelt werden. Diese ermöglichen einen engen inhaltlichen Austausch und eine bestmögliche Patientenversorgung. Ein besonderes Merkmal der Region ist das PH-Netzwerk Nordrhein, welches überregionale Qualitätsstandards unter den PH-Zentren der Region gewährleistet.
Beitrag für die Zeitschrift „Lunge im Fokus“
Simon Herkenrath, Stephan Rosenkranz
Diagnostischer Algorithmus bei pulmonaler Hypertonie angelehnt an den Vorschlag der internationalen Arbeitsgruppe „Diagnosis of pulmonary hypertension“ des 6th World Symposium on Pulmonary Hypertension (Nizza 2018).
Simonneau ERJ 2019 53: 1801913
Aktuelle klinische Klassifikation der pulmonalen Hypertonie nach Nizza 2018
- PAH
- Idiopathische PAH
- Hereditäre PAH
- Medikamenten- und Toxininduzierte PAH
- PAH assoziiert mit:
- Bindegewebserkrankung
- HIV-Infektion
- Portale Hypertension
- Angeborene Herzfehler
- Schistosomiasis
- PAH Langzeit-Responder auf Kalziumkanalblocker
- PAH mit Merkmalen einer Pulmonalvenenbetieligung (PVOD / PCH)
- Persistierende PH des Neugeborenen
- PH aufgrund einer Erkrankung des linken Herzens
- PH aufgrund von Linksherzinsuffizienz mit erhaltener LVEF
- PH aufgrund von Linksherzinsuffizienz mit reduzierter LVEF
- Herzklappenerkrankungen
- Angeborene / erworbene kardiovaskuläre Erkrankungen mit resultierender postkapillären PH
- PH aufgrund von Lungenerkrankungen und / oder Hypoxie
- Obstruktive Lungenerkrankung
- Restriktive Lungenerkrankung
- Andere Lungenerkrankung mit gemischtem restriktivem / obstruktivem Muster
- Hypoxie ohne Lungenerkrankung
- Entwicklungsstörungen der Lunge
- PH aufgrund einer Lungenarterienobstruktion
- Chronisch-thromboembolische PH
- Andere Lungenarterienobstruktionen
- PH mit unklaren und / oder multifaktoriellen Mechanismen
- Hämatologische Erkrankungen
- Systemische und Stoffwechselstörungen
- Andere Erkrankungen
- Komplexe angeborene Herzkrankheiten
Simonneau ERJ 2019 53: 1801913
Hämodynamische Definitionen der pulmonalen Hypertonie

Galiè European Heart Journal, Volume 37, Issue 1, 1 January 2016, Pages 67–119
Risikostratifizierung bei pulmonalarterieller Hypertonie nach Galiè, ESC Guideline 2015